
More Information
Submitted: August 08, 2024 | Approved: August 23, 2024 | Published: August 26, 2024
How to cite this article: De Cagna MR, Notaristefano N, Schiavone M, Palatella G, Ranù R, Presicci C, et al. Autoantibodies in Autoimmune Addison’s Disease: Why are they Important? Arch Pathol Clin Res. 2024; 8(1): 012-015. Available from: https://dx.doi.org/10.29328/journal.apcr.1001042.
DOI: 10.29328/journal.apcr.1001042
Copyright License: © 2024 De Cagna MR, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Grade; Immunohistochemistry; Meningioma; Mitotic index; pHH3

Autoantibodies in Autoimmune Addison’s Disease: Why are they Important?
Maria Rosaria De Cagna1#, Norma Notaristefano1#, Maurizio Schiavone2, Gianluca Palatella1, Federica Ranù1, Carmela Presicci1, Valerio Cecinati2 and Marilina Tampoia1*
1U.O.C. Patologia Clinica, Ospedale SS. Annunziata, Taranto, Italy
2U.O.C. Pediatria, Ospedale SS. Annunziata, Taranto, Italy
#Contributed equally
*Address for Correspondence: Marilina Tampoia, U.O.C Patologia Clinica, Ospedale SS. Annunziata, Taranto, Italy, Email: marilina.tampoia@asl.taranto.it
Primary adrenocortical insufficiency or Addison’s disease (AD) is a rare, life-threatening condition with different aetiologies, but the most common cause is autoimmune destruction of the adrenal cortex. Autoimmune Addison’s disease (AAD) can present as an isolated condition or associated with others, as part of an autoimmune polyglandular syndrome (APS). The aim of this work is to investigate and emphasise the roles of autoantibodies in adrenocortical insufficiency, through the description of three clinical cases.
Primary adrenocortical insufficiency or Addison’s disease (AD) is a rare condition, which occurs in the general population with a frequency of 1 in 8000 individuals approximately [1]. The diagnosis is often complicated by non-specific signs and symptoms at clinical onset, therefore, it is a potentially life-threatening condition, if not recognized and promptly treated [2].
The most common etiology is the autoimmune destruction of the adrenal cortex, which has an external-internal cytoarchitecture as follows: pars glomerulosa, responsible for the production of mineralocorticoid hormones (mainly aldosterone) regulated by renin-angiotensin system; pars fasciculata, which occupies the major part of the cortex, responsible for the synthesis of glucocorticoid hormones (mainly cortisol) under the control of ACTH; pars reticularis responsible for the synthesis of androgens (dehydroepiandrosterone, delta-4-androstenedione, testosterone) [3].
The natural history of autoimmune Addison’s disease shows five sequential stages of cortical function impairment, based on the response to challenge test with exogenous ACTH in patients with autoantibody positivity [4]. Stage 0 defines a potential or latent condition, characterized by autoantibody positivity and normal adrenal function. Stages 1,2 and 3 are subclinical ones and show an increasing impairment of function, highlighted by mineralocorticoid and glucocorticoid hormone assays. Stage 4 defines clinical hypoadrenalism, as characterized by very high ACTH levels, very low cortisol values, and the appearance of clinical symptoms (asthenia, abdominal pain, hypotension, hyponatremia, and hyperkalemia) [5].
Autoimmune Addison’s disease (AAD) can finally present as an isolated condition or associated with other pathologies, in autoimmune polyglandular syndromes: type 1 (APS-1), type 2 (APS-2), and type 4 (APS-4), which identify immunological disorders characterized by the coexistence of two or more clinically manifest autoimmune diseases and with an endocrine pathology at onset [6-8].
The aim of this work is to highlight, through the description of three clinical cases, the roles of circulating anti-adrenal cortex autoantibodies (ACA, adrenal cortex autoantibodies) and anti-21-hydroxylase (anti-21-OH) autoantibodies, in autoimmune Addison’s disease
Clinical case I
A male patient, aged 16, arrived in the emergency room due to severe asthenia, hypotension, fever, and dehydration. He was admitted to the Pediatric Department and subjected to haematochemical and instrumental investigations. Laboratory tests showed dyselectrolytemia, with hyponatremia (Sodium: 109 mEq/L-r.i. 130-150) and hyperkalemia (Potassium: 6.4 mEq/L – r.i. 3.5-5.2). Serological and molecular tests denied a possible infectious etiology; Computed Tomography scan and Magnetic Resonance Imaging excluded space-occupying lesions in the brain.
In the suspicion of adrenocortical insufficiency ACTH, cortisol, renin and aldosterone were measured and showed an increase in ACTH and renin levels (ACTH >1250 pg/mL - r.v. <46; renin >500 μIU/mL – r.i. 2.8-39.9), hypocortisolemia (cortisol 2.3 μg/dL – r.i. 5-27) and normal aldosterone value (75.81 pg/mL – r.i. 30-160). ACTH stimulation test confirmed adrenal insufficiency and the patient was immediately started on therapy with hydrocortisone and fludrocortisone.
An indirect immunofluorescence method (IFI) on primate adrenal tissue (IFA Adrenal Cortex, Euroimmun, Germany) was used, according to the manufacturer’s instructions, to search for circulating autoantibodies against the adrenal cortex and showed a positive result: the fluorescent signal affected the entire microscopic section including pars glomerulosa and fasciculata (Figure 1). The autoantibody positivity was confirmed by an immunoenzymatic method (21-Hydroxylase Autoantibody ELISA Kit, RSR, United Kingdom) which detected autoantibodies against 21-hydroxylase, an enzyme involved in adrenocortical steroidogenesis and a specific target of anti-adrenal autoantibodies [9]. This test showed high levels of anti-21-OH autoantibodies (2.2 units/mL, r.v. < 0.4). These findings led to the etiological diagnosis of Autoimmune Addison’s disease at clinical stage 4.
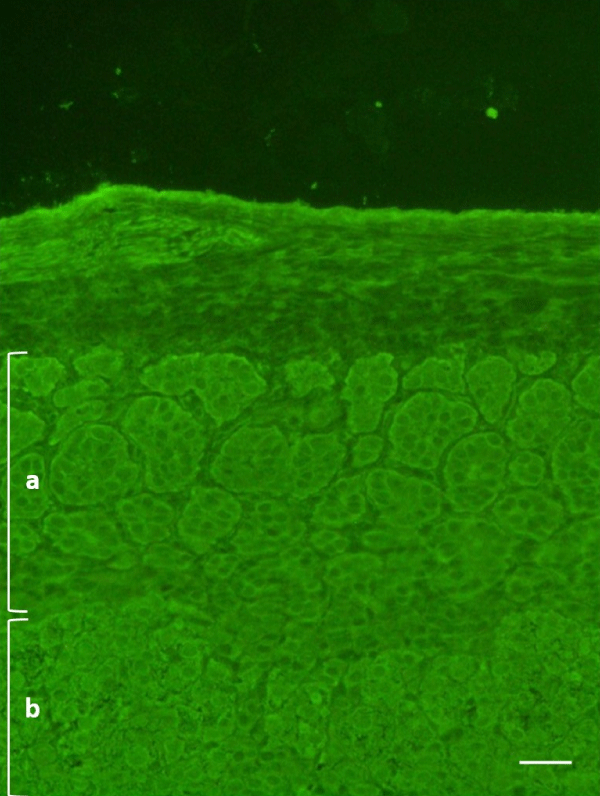
Figure 1: Indirect immunofluorescence on primate adrenal tissue (dilution 1:5, 400x) – case I. a: pars glomerulosa; b: pars fasciculata. Scale bar: 100 μm
Clinical case II
A male patient, 17 years old, with a previous diagnosis of Hashimoto’s thyroiditis and type I diabetes mellitus, was admitted to the Pediatric Department for severe dehydration, asthenia, and hypotension. Preliminary laboratory tests showed dyselectrolytemia, with hyponatremia (Sodium: 114 mEq/L-r.i. 130-150) and hyperkalemia (Potassium: 5.9 mEq/L – r.i. 3.5-5.2). Serological and molecular tests denied a possible infectious etiology, so in the suspicion of adrenocortical insufficiency, the clinician proceeded with the request for further tests which revealed very high levels of ACTH and Renin (ACTH >1250 pg/mL - r.v. <46; Renin >500 μIU/mL – r.i. 2.8-39.9), reduction of cortisolemia (cortisol 3.5 μg/dL – r.i. 5-27) and normal value of aldosterone (2.97 ng/dL – r.i. 1.76-23.2). The patient was started on therapy with hydrocortisone and fludrocortisone. In order to better investigate the etiology of adrenocortical insufficiency, ACA was searched by an IFI method (IFA Adrenal Cortex, Euroimmun, Germany) with positive results (Figure 2) and 21-OH autoantibodies, subsequently evaluated (21-Hydroxylase Autoantibody ELISA Kit, RSR, United Kingdom), confirmed the autoimmune etiology. Because of concomitant Hashimoto’s thyroiditis, type I diabetes mellitus, and Autoimmune Addison’s disease at the clinical stage, the patient was discharged with the diagnosis of APS-2.
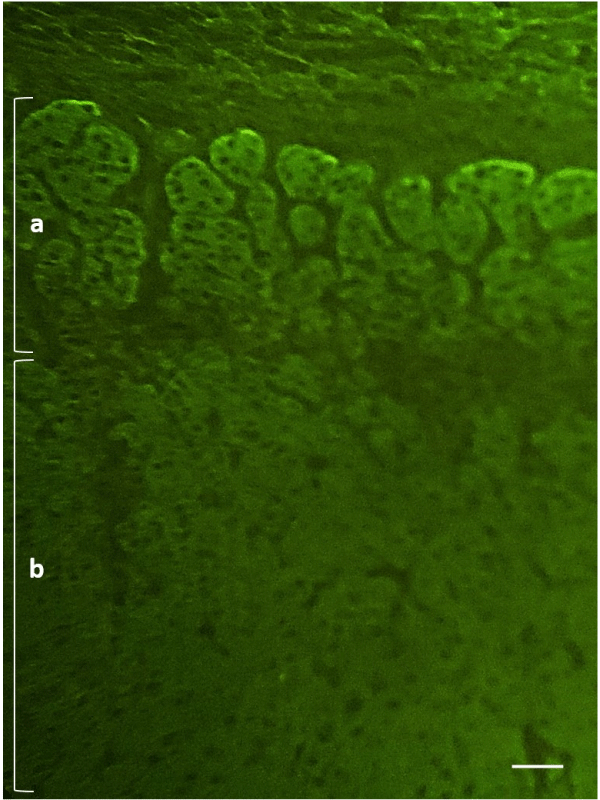
Figure 2: Indirect immunofluorescence on primate adrenal tissue (dilution 1:5, 400x) – case II. a: pars glomerulosa; b: pars fasciculata. Scale bar: 100 μm
Clinical case III
A female patient, 14 years old, was admitted to the Pediatric Department for follow-up of autoimmune polyendocrine syndrome type 1 (APS-1), also known as APECED (Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy), diagnosed in 2016 due to the association of mucocutaneous candidiasis, hypoparathyroidism and the presence of mutations in the AIRE gene (AutoImmune REGulator gene).
Due to the young age, personal clinical history, and the awareness that Autoimmune Addison’s disease can be associated with mucocutaneous candidiasis and chronic hypoparathyroidism in APS-1, despite the lack of adrenal insufficiency clinical manifestations, ACTH, cortisol, and aldosterone were assayed and were within reference limits (ACTH 19.7 pg/mL –r.v. < 46; cortisol 12.9 μg/dL –r.i. 5-27; aldosterone 121.3 pg/mL –i.r. 30 -160). The search for anti-adrenal cortex autoantibodies by an IFI method (IFA Adrenal Cortex, Euroimmun, Germany) was instead positive (Figure 3) and the enzyme immunoassay (21-Hydroxylase Autoantibody ELISA Kit, RSR, United Kingdom) for detection of anti-21-OH autoantibodies showed a weak positivity (0.6 units/mL, r.v. <0.4).
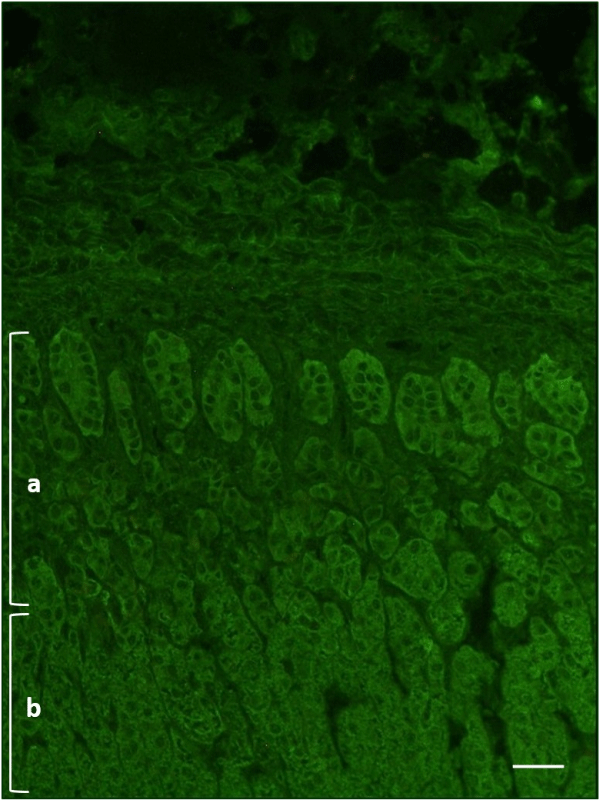
Figure 3: Indirect immunofluorescence on primate adrenal tissue (dilution 1:5, 400x) – case III. a: pars glomerulosa; b: pars fasciculata. Scale bar: 100 μm
The patient, with known APECED syndrome, was therefore discharged with the diagnosis of latent Addison’s disease, on the basis of normal adrenal function and specific autoantibody positivity, with the indication for clinical and laboratory follow-up, also including the adrenal hormones assays.
Three cases with positive ACA confirmed with the enzyme immunoassay for anti-21-OH, have been described (Table 1).
| Table 1: Main features of the clinical cases. | |||
| Case I | Case II | Case III | |
| Age | 16 | 17 | 14 |
| Sex | Male | Male | Female |
| Remote medical history | Nothing to report | -Hashimoto's thyroiditis -Type 1 Diabetes Mellitus |
|
| ACTH | >1250 pg/mL (r.v. <46) | >1250 pg/mL (r.v. <46) | 19,7 pg/mL (r.v. <46) |
| Cortisol | 2,3 μg/dL (r.i. 5-27) | 3.5 μg/dL (r.i. 5-27) | 12,9 μg/dL (r.i. 5-27) |
| ACA (IFI) | Positive | Positive | Positive |
| Anti-21-OH | Positive | Positive | Positive |
| ACA: adrenal cortex autoantibodies; Anti-21-OH: anti-21-hydroxylase autoantibodies; IFI: indirect immunofluorescence; APECED: Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy | |||
Since the probability of an accurate diagnosis of autoimmune Addison’s disease is greater than 99% in the case of double positivity for ACA and for anti-21-OH, it was decided to carry out an ACA screening in IFI and confirm the outcome in immunoenzymatic [10].
In the natural history of autoimmune Addison’s disease the time between the detection of specific autoantibodies, the appearance of adrenal functional modifications, and the evidence of symptoms is variable and and can even last a few years [11]. The cases described are at the opposite ends of the natural history stadiation [4,5].
In the first case, ACA and anti-21-OH antibodies result in excellent diagnostic markers in the etiological classification of autoimmune adrenocortical insufficiency [9]. In fact, they are detectable in more than 90% of patients with Addison’s disease at clinical onset, although they often appear even before the development of full-blown adrenal dysfunction [6,9], which becomes evident only when 70% of the cortex is damaged. The Autoimmunology laboratory’s readiness to perform an immunofluorescence test and an enzyme immunoassay allowed the diagnostic and etiological classification in a rather short time.
In the second case, the positivity for ACA and anti-21-OH autoantibodies is important not only to define the autoimmune etiology of Addison’s disease but also to allow the diagnosis of APS-2, an autoimmune polyendocrine syndrome characterized by the compulsory presence of Addison’s disease, associated with autoimmune thyroiditis and type 1 diabetes mellitus.
In the third case, it is clear that positivity for ACA and anti-21-OH autoantibodies have an important predictive value, especially in patients with autoimmune polyendocrine syndromes already diagnosed, even in the absence of adrenocortical insufficiency clinical manifestations. In APECED patients without clinically evident Addison’s disease, a positivity for ACA and anti-21-OH autoantibodies allows us to predict the full-blown pathology within ten years in almost all cases9. It contributes to supporting the strong association between the presence of ACA and anti-21OH and the development of autoimmune Addison’s disease [7].
In two of the three cases, ACTH and Cortisol levels were outside the normal ranges. These findings have been very important in leading to the diagnosis of adrenal insufficiency. ACA and anti-21-OH positivity, subsequently evaluated, led to the identification of the autoimmune etiology of this diagnosis.
In the third case, the diagnostic course for Addison’s disease started with ACA and anti-21-OH positivity. ACTH and Cortisol levels assayed afterwards, have been found within normal ranges, leading to the diagnosis of a latent autoimmune Addison’s disease.
In summary, the three cases outline the heterogeneity of autoimmune Addison’s disease natural history and clearly describe the key roles in this context of anti-adrenal autoantibodies.
Ethical declarations
All procedures described in the study were implemented in accordance with the ethical standards established by the 1964 Helsinki Declaration and subsequent amendments. Informed consent of the minor patients’ parents was obtained.
- Saverino S, Falorni A. Autoimmune Addison's disease. Best Pract Res Clin Endocrinol Metab. 2020; 34(1):101379. Available from: https://doi.org/10.1016/j.beem.2020.101379
- Puttanna A, Cunningham AR, Dainty P. BMJ Case Rep. 2013; bcr-2013-010473. Available from: https://doi.org/10.1136/bcr-2013-010473
- Abdellatif AB, Fernandes-Rosa FL, Boulkroun S and Zennaro MC Vascular and hormonal interactions in the adrenal gland. Front. Endocrinol. 2022; 13:995228. Available from: https://doi.org/10.3389/fendo.2022.995228
- Betterle C, Garelli S, Presotto F, Furmaniak J. From appearance of adrenal autoantibodies to clinical symptoms of Addison’s disease: natural history. Front Horm Res 2016; 46:133–145. Available from: https://doi.org/10.1159/000443872
- Manso J, Pezzani R, Scarpa R, Gallo N, Betterle C. The natural history of autoimmune Addison's disease with a non-classical presentation: a case report and review of literature Clin Chem Lab Med 2018; 56(6):896-900. Available from: https://doi.org/10.1515/cclm-2017-1108
- Betterle C, Dal Pra C, Mantero F, Zanchetta R. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr Rev 2002; 23:327–64. Available from: https://doi.org/10.1210/edrv.23.3.0466
- Betterle C, Zanchetta R. Update on autoimmune polyendocrine syndromes (APS). Acta Biomed 2003; 74:9–33 PMID: 12817789. Available from: https://pubmed.ncbi.nlm.nih.gov/12817789/
- Sorrentino MC, Imbastaro T, Tozzoli R. Le sindromi poliendocrine autoimmuni. In: Tozzoli R, Bizzaro N, Villalta D, Tonutti E. Il laboratorio nelle malattie autoimmuni d’organo. Esculapio 2019; 155-178.
- Muraro V, Deleonardi G, Bizzaro N. Serological markers of autoimmune Addison disease RiMeL 2023;19(3):157-61.
- Falorni A, Laureti S, De Bellis A, Zanchetta R, Tiberti C, Arnaldi G, et al.; SIE Addison Study Group. Italian addison network study: update of diagnostic criteria for the etiological classification of primary adrenal insufficiency. J Clin Endocrinol Metab 2004; 89:1598–604. Available from: https://doi.org/10.1210/jc.2003-030954
- Teti C, Tozzoli R, Bagnasco M. Gli autoanticorpi nelle malattie autoimmuni del surrene. In: Tozzoli R, Bizzaro N, Villalta D, Tonutti E. Il laboratorio nelle malattie autoimmuni d’organo. Esculapio 2019; 127-132